How Neural Networks Can Use Quantum Mechanics to Treat Epilepsy
For the millions of people with drug-resistant epilepsy, new treatments are a critical need. This article explores how a new AI tool, OrbMol, is able to use quantum mechanics to simulate the complex molecular interactions at the heart of the disease.
OrbMol and the Rise of AI in Materials and Molecular Modelling
The properties of every material are dictated by its atomic structure and molecular dynamics. For decades, predicting a material's properties meant running slow, computationally expensive quantum simulations. Now, AI is revolutionising this field by delivering the same high-fidelity predictions in a fraction of the time, allowing scientists to design new materials on a computer instead of through costly lab-based trial and error.
To lead this change, Orbital Materials has released the Orb family, a suite of state-of-the-art, open-source AI models designed for the rapid and accurate simulation of advanced materials across a vast number of applications, from energy storage to aerospace to the future of microchips. The latest addition is OrbMol, a neural network designed specifically for complex molecular systems like metal complexes, biomolecules, and electrolytes. With this capability, OrbMol unlocks new possibilities for researchers tackling challenges in the pharmaceutical and biotechnology industries.
Let’s now examine the capabilities of OrbMol using epilepsy drug development as a case study to validate its performance in simulating a complex biological target.
A New Pathway for Drug-Resistant Epilepsy
Epilepsy affects around 50 million people. Most anti-seizure medicines control seizures by directly modifying electrical signalling within neurons or their ion channels. However, around a third of people are resistant to these standard treatments. For this group, alternative approaches are needed. Carbonic anhydrase (CA) enzyme inhibitors offer a new pathway for seizure medicines for this group resistant to standard treatment.
The CA enzyme helps regulate the pH, which controls neuronal excitability within the brain by catalysing the reversible reaction of CO₂ ⇄ bicarbonate. At the core of the CA enzyme is a zinc ion. This zinc ion enables the enzyme to accelerate the CO₂ ⇄ bicarbonate reaction to be fast enough to tightly regulate pH and CO₂ levels during neuronal activity.
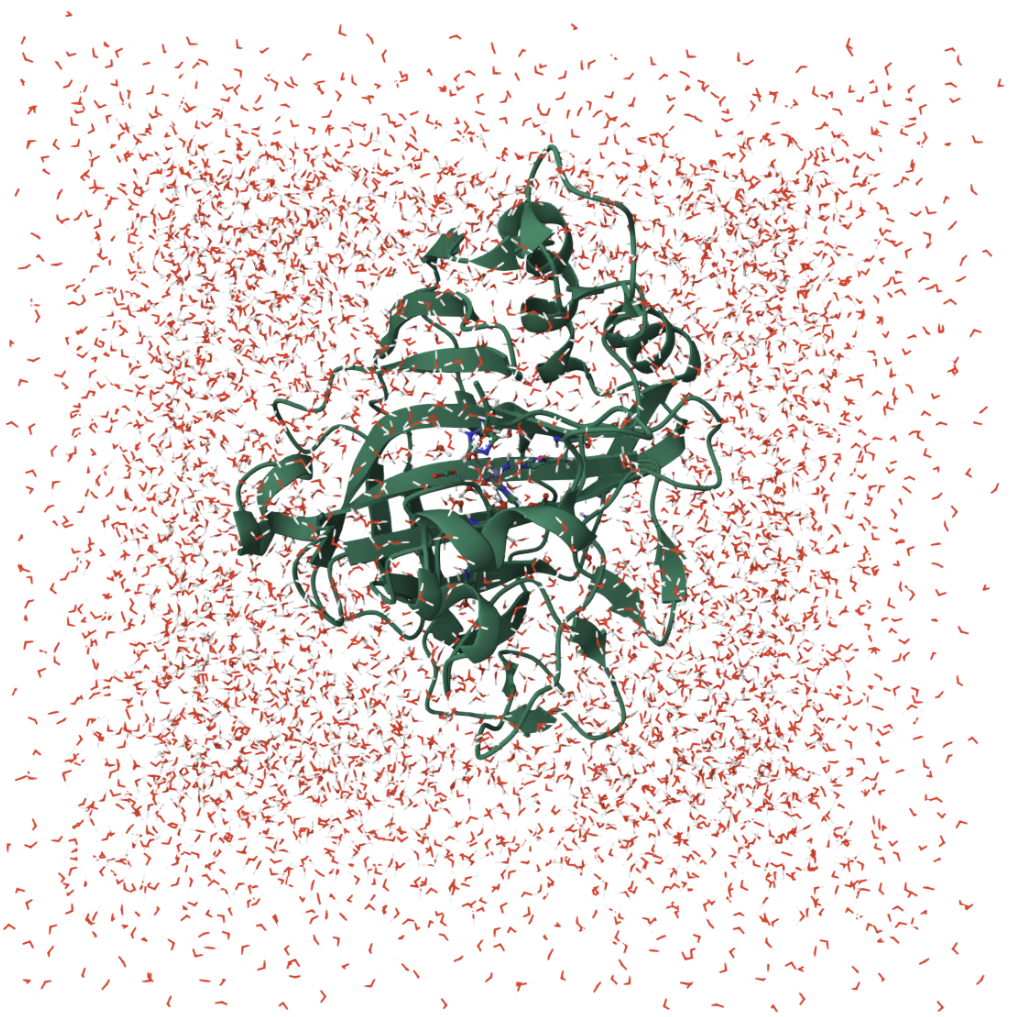
The alternative anti-seizure CA inhibitor drug binds to the zinc ion and blocks the CO₂ ⇄ bicarbonate reaction, which subtly shifts the local pH to become more acidic. This acidification, in turn, reduces neuronal excitability, calming the hyperactive firing that drives seizures.
So, we’ve established how the CA enzyme and its zinc centre are central to how these inhibitor drugs work. To develop effective treatments, we need to be able to model, simulate, and predict the whole enzyme and its interactions accurately.
This brings us to OrbMol's approach and its effectiveness.
OrbMol: The Innovation and the Validation
A Data-Driven Model Conditioned on Physical Priors
Traditional models have often struggled with metal coordination and the subtle electronic effects that a zinc ion would bring into the equation. OrbMol has been built to overcome this in two ways.
Firstly, it was trained on the Open Molecules 2025 (OMol25) dataset, which consists of over 100 million DFT calculations at the highly accurate ωB97M-V/def2-TZVPD level of theory. This dataset incorporated large, charged, and magnetic systems, which capture many of the electronic and structural complexities also present in our CA enzyme.
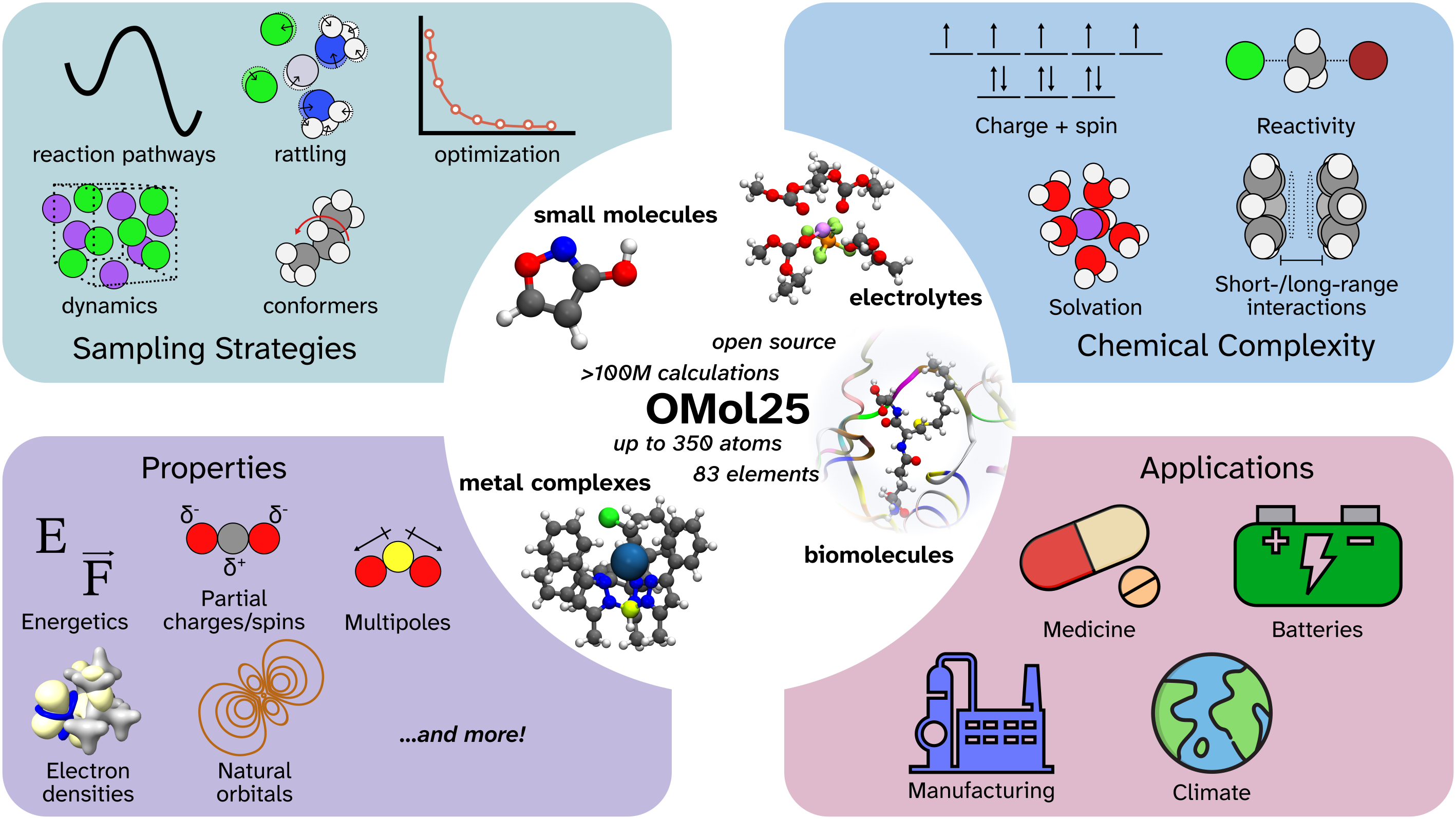
Secondly, OrbMol further enhances accuracy for the charged and spin-polarized systems found in the CA enzyme through specifically conditioning on total charge and spin multiplicity. This means the model factors in these variables when predicting how a molecule behaves.
With this conditioning, Orbmol isn't just told the atom and the atomic position, it is also told:
“This is a zinc ion, charge = +2, multiplicity = singlet”
Without this context, the geometry of the active site could look “reasonable,” but the electronic structure would be wrong, leading to incorrect predictions of:
- The Binding energies for inhibitors, critical for screening and optimising drug candidates.
- The pKa shifts in the water/hydroxide, which control how readily the enzyme activates water for catalysis.
- The Proton transfer dynamics that the enzyme’s activity depend on.
Engineered for Speed and Efficiency
In drug development, the speed of simulation is critical: faster models allow researchers to explore more of the chemical landscape, shorten the design–test–learn feedback loop, and run cheaper calculations, which is particularly valuable for labs without access to large-scale computing resources.
OrbMol was specifically engineered for speed, with an architecture optimised for GPUs. This design allows OrbMol to match the performance of state-of-the-art universal machine learning potentials UMA and eSEN while being at least twice as fast, offering a significant efficiency advantage.
Pushing the Model to its Limits
The GMTKN55, PLA15, and Wiggle150 benchmarks are three crucial tests designed to stress-test the accuracy and robustness of molecular modelling methods. Each assesses a different aspect of performance crucial for accurately modelling our CA enzyme and its interactions.
GMTKN55: The Gold Standard Test for Chemical Accuracy
The GMTKN55 benchmark comprises 55 tests benchmarked against high-level quantum chemistry reference data, evaluating computational methods across thermochemistry, reaction kinetics, and noncovalent interactions. For the CA enzyme, this is vital for correctly predicting the binding energies and reaction dynamics of potential inhibitor drugs.
PLA15: Does It Scale? Testing on Realistic Systems
While GMTKN55 tests a model’s fundamental accuracy on smaller systems (10–50 atoms), the PLA15 benchmark measures protein–ligand interaction energies for 15 large complexes (600–2000 atoms). This allows evaluation of accuracy on systems too large for direct quantum calculations, bridging the gap between small-molecule benchmarks and realistic drug design targets. Essentially, this benchmark confirms that the model's accuracy scales up to realistic biological targets like the CA enzyme.
Wiggle150: The Perfect Fit: Modeling Strained Shapes
The Wiggle150 benchmark is crucial because it tests a model's accuracy on strained, non-equilibrium molecular shapes. This is directly relevant to our CA inhibitor, which may be stable on its own but must contort into a specific, and often energetically unfavourable, conformation to bind effectively to the enzyme's active site. Predicting this strained shape correctly is essential for identifying effective drug that actually works in practice.
The Results
OrbMol excels across all three benchmarks. This exceptional performance validates its predictions for the CA enzyme (from binding strength to catalytic behaviour), showing they can be reliably applied to real-world drug development. Accurately predicting these factors is often the difference between a promising compound and a clinical dead end.
For a detailed analysis of OrbMol’s performance on these benchmarks, see the full technical paper here.
Putting Theory into Practice: The CA Enzyme Simulation
OrbMol was used to simulate our CA enzyme, a system of over 20,000 atoms including the crucial coordinated zinc ion, for more than 230 ps without constraints. This demonstrates that OrbMol can handle large, complex biological systems under realistic conditions.
The resulting structure remained stable relative to the original PDB structure, showing that OrbMol can maintain the enzyme’s native conformation over long simulations without artificial drift. The simulation also achieved a very low RMSD of 0.6 Å, indicating a high level of accuracy in reproducing the experimentally determined structure. For context, simulations using classical forcefields typically show RMSDs exceeding 2 Å over similar timescales.
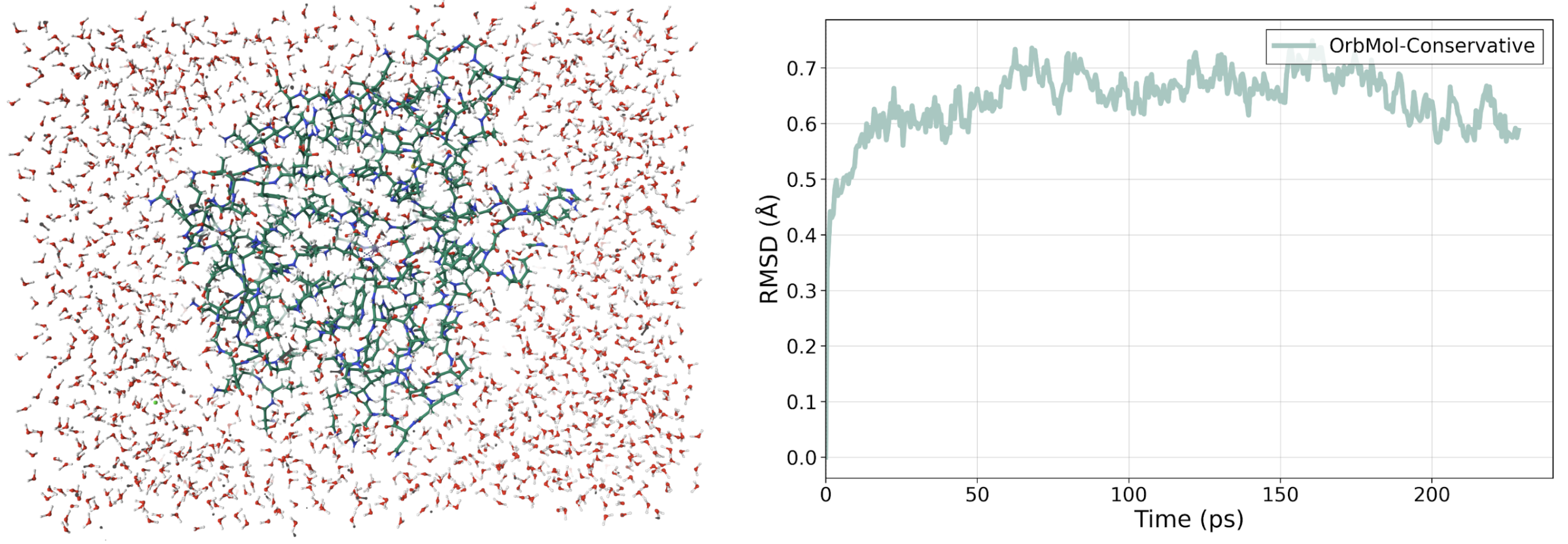
OrbMol then went further to model the key interactions within the CA enzyme, the same ones that underpin the development of effective treatments for epilepsy. In the simulation, a CO₂ molecule randomly placed in the enzyme’s active site quickly displaced two water molecules and settled into the known binding site next to the zinc, surrounded by hydrophobic residues. This behaviour had not been observed with earlier Orb models and shows that OrbMol can accurately capture the weak physisorption and dispersion interactions that drive this kind of binding.

Closing statements
These results show that OrbMol is both accurate and practical for simulating complex biological systems like carbonic anhydrase. Its combination of speed, accuracy, and scalability makes it a powerful tool for advancing drug discovery and development.
While this article focused on a pharmaceutical challenge, the implications of OrbMol are far broader. Ultimately, tools like Orbmol represent the future of molecular and materials science. This technology signals a paradigm shift away from slow, lab-based trial and error to an era of rapid, in silico creation. The path from an atomic blueprint to a revolutionary new material, whether it's a life-saving drug, a more efficient battery, or a stronger aerospace alloy, will now be navigated through precise, AI-driven design. This accelerates innovation across every industry, empowering scientists to build the foundational materials for the next generation of technology.
Articles